Research
The publications and manuscripts listed here are those affiliated with CompBioClub. For complete lists, see the members' scholar pages.
-
 Synergistic Inhibition of Notch Signaling and Forced Cell Cycle Re-entry Drive Müller Glia Reprogramming in Uninjured Mouse RetinaBaoshan Liao, Chengshang Lyu, Yuqing Jiang, Shanggong Liu, Waiho Wong, Jiadong Zhang, Hoyin Tsang, Junxi Xie, Lingxi Chen, Qinrong Zhang, and Wenjun XiongeLife, 2026
Synergistic Inhibition of Notch Signaling and Forced Cell Cycle Re-entry Drive Müller Glia Reprogramming in Uninjured Mouse RetinaBaoshan Liao, Chengshang Lyu, Yuqing Jiang, Shanggong Liu, Waiho Wong, Jiadong Zhang, Hoyin Tsang, Junxi Xie, Lingxi Chen, Qinrong Zhang, and Wenjun XiongeLife, 2026In regenerative species, such as teleost fish, Müller glia (MG) autonomously re-enter the cell cycle after injury and give rise to functional retinal neurons. In contrast, the loss of retinal neurons in mammals is irreversible due to the limited proliferative and regenerative ability of MG. Various strategies have been developed to induce proliferation of mature mouse MG with or without injury, yet most MG daughter cells retain glial cell fate. Here, we found that MG progenies maintain high Notch signaling, which may constrain their neurogenic potential. Conditional deletion of Rbpj, the central transcriptional effector of Notch, induced limited MG-to-neuron conversion in mature MG without proliferation. However, Rbpj deletion, combined with forced MG proliferation by overexpressing cyclin D1 and suppressing p27Kip1, significantly promoted MG dedifferentiation and ectopic expression of the neuronal marker Otx2 in MG daughter cells in uninjured mouse retina. Combining Notch inhibition with MG cell cycle re-activation not only increased the numbers of bipolar- and amacrine-like cells generated from MG but also promoted the further differentiation toward ON-cone, OFF-cone, and rod-bipolar subtypes. Single-nucleus RNA and ATAC sequencing data revealed that Notch inhibition facilitated the formation of MG-derived progenitor-like cells while MG proliferation increased chromatin accessibility of neurogenic genes. Notably, most MG-derived cells survived long term despite incomplete maturation. Together, our findings delineate how Notch inhibition and MG proliferation, alone or in combination, influence the regenerative potential of MG in the mammalian retina.
@article{liao2026synergistic, title = {Synergistic Inhibition of Notch Signaling and Forced Cell Cycle Re-entry Drive M{\"u}ller Glia Reprogramming in Uninjured Mouse Retina}, author = {Liao, Baoshan and Lyu, Chengshang and Jiang, Yuqing and Liu, Shanggong and Wong, Waiho and Zhang, Jiadong and Tsang, Hoyin and Xie, Junxi and Chen, Lingxi and Zhang, Qinrong and Xiong, Wenjun}, journal = {eLife}, volume = {15}, year = {2026}, publisher = {eLife Sciences Publications Limited}, doi = {10.7554/eLife.111251.1}, peerreviewed = {true}, member = {Lyu, Chengshang}, topic = {Retina, scRNA} } -
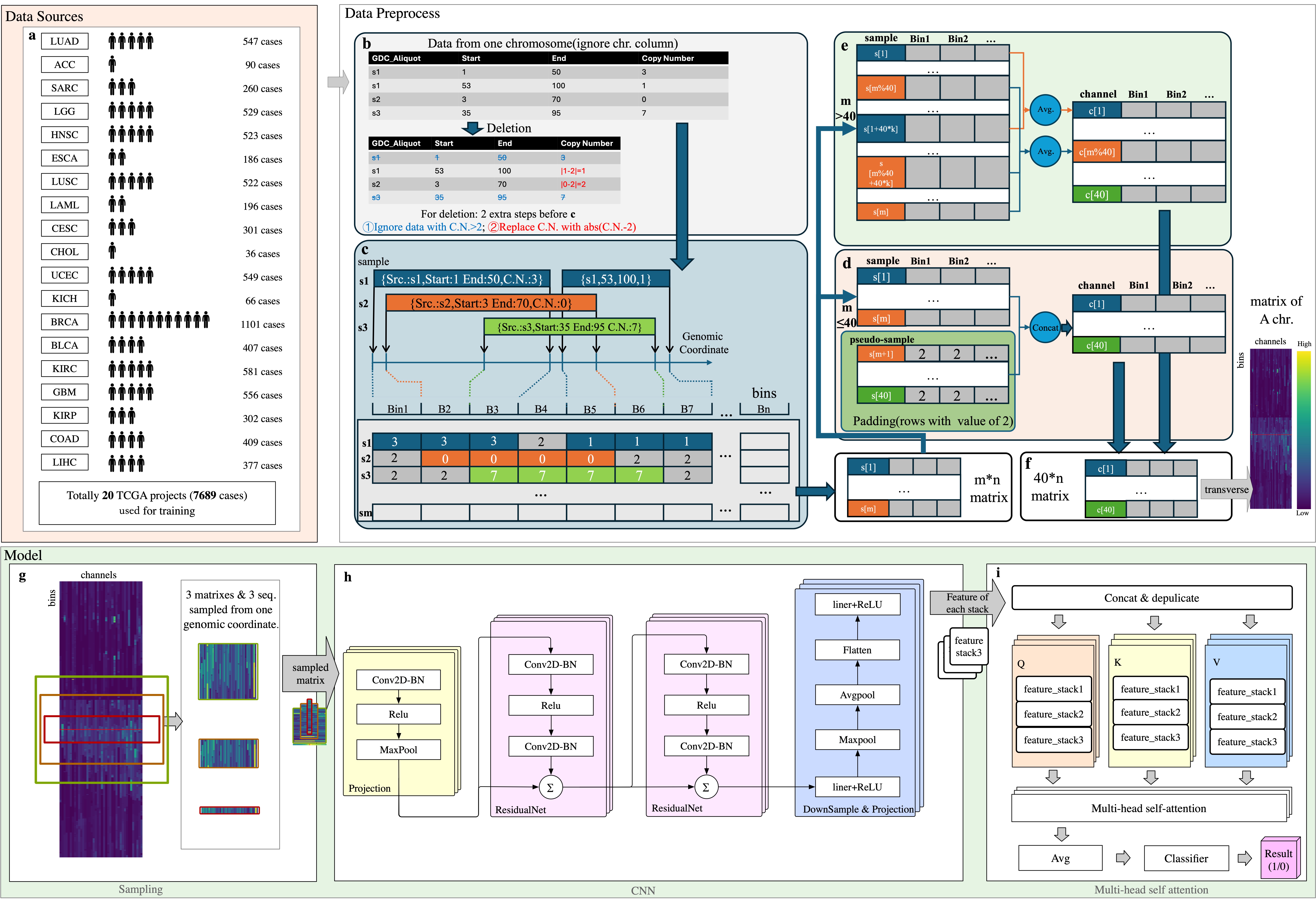 CRESCENT: A Deep Learning Framework with Multi-Scale Attention for Detecting Recurrent Copy Number AberrationsXikang Feng†*, Zheng Xu†, Sisi Peng, Jieyi Zheng, Chuan Ma, Qiangguo Jin*, and Lingxi Chen*Briefings In Bioinformatics, 2026
CRESCENT: A Deep Learning Framework with Multi-Scale Attention for Detecting Recurrent Copy Number AberrationsXikang Feng†*, Zheng Xu†, Sisi Peng, Jieyi Zheng, Chuan Ma, Qiangguo Jin*, and Lingxi Chen*Briefings In Bioinformatics, 2026Recurrent copy number alterations (CNAs) are fundamental drivers of tumorigenesis, yet identifying them reliably remains a challenge due to the extreme variability in their genomic scale and context. Current methods often struggle to balance sensitivity across focal, segmental, and arm-level events. Here, we present CRESCENT, a deep learning framework designed to detect recurrent CNAs by integrating multi-scale sampling with convolutional neural networks and self-attention mechanisms. By processing copy number profiles from 7,689 cases across 20 TCGA cancer projects, CRESCENT learns to distinguish recurrent drivers from background noise through parallel feature fusion. In rigorous leave-one-project-out cross-validation, the model demonstrated robust generalization, achieving AUCs of 0.894-0.967 for amplifications and 0.804-0.929 for deletions in representative cohorts (BLCA, SARC, GBM, UCEC). Finally, extending beyond the TCGA-specific cross-validation, we trained a unified pan-cancer model to assess CRESCENT’s generalizability on simulated datasets and independent, non-TCGA cancer cohorts (CGCI and TARGET). Benchmarking against standard tools, including GISTIC2 and RUBIC, reveals that CRESCENT offers superior detection balance, identifying the highest total number of significant events across focal and broad scales. Moreover, extensive focal gene expression validation and pathway annotation, coupled with survival analysis, highlights that CRESCENT identifies critical oncogenic drivers and prognostic markers that conventional statistical methods often overlook. In all, CRESCENT provides a highly sensitive, generalized approach for decoding tumor evolution.
@article{CRESCENT, title = {CRESCENT: A Deep Learning Framework with Multi-Scale Attention for Detecting Recurrent Copy Number Aberrations}, author = {Feng, Xikang and Xu, Zheng and Peng, Sisi and Zheng, Jieyi and Ma, Chuan and Jin, Qiangguo and Chen, Lingxi}, journal = {Briefings In Bioinformatics}, pages = {2026--03}, year = {2026}, publisher = {Oxford University Press}, doi = {10.1093/bib/bbag167}, peerreviewed = {true}, topic = {CNA, AI, Cancer} } -
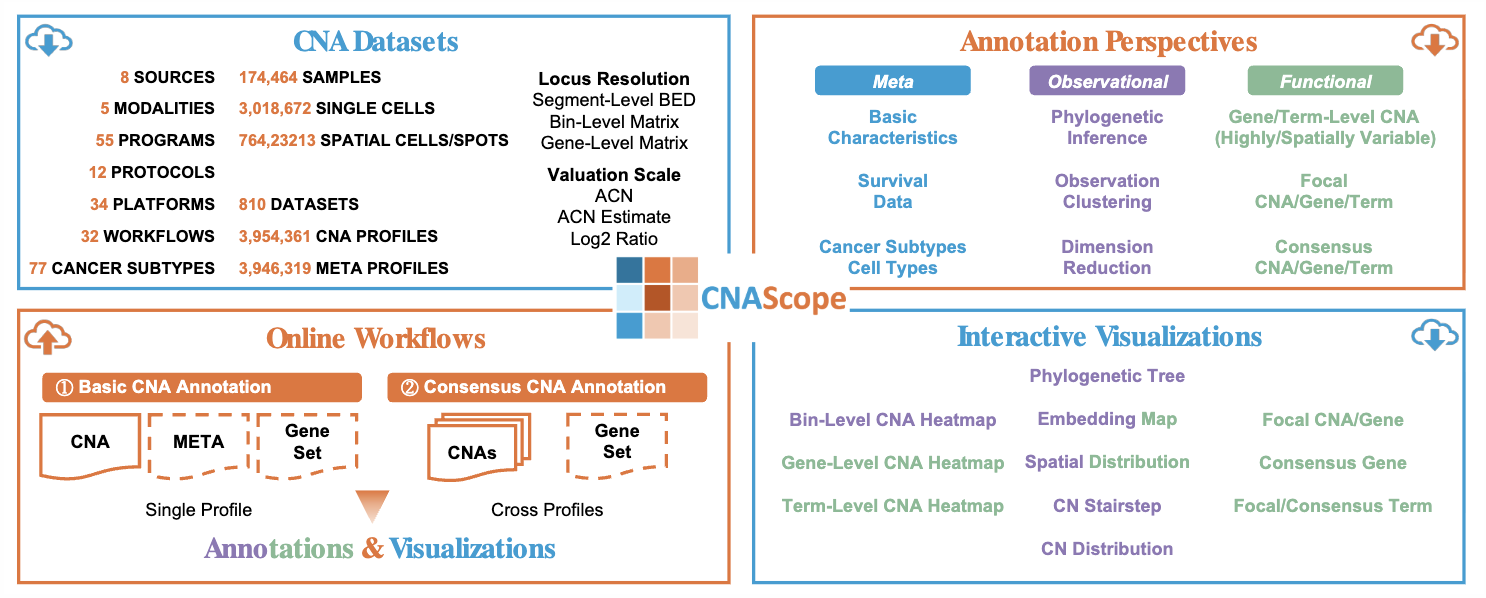 CNAScope: Pan-Cancer Copy Number Aberration Database with Functional Annotation and Interactive VisualizationXikang Feng†*, Jieyi Zheng†, Sisi Peng†, Anna Jiang†, Ka Ho Ng, Chengshang Lyu, Qiangguo Jin*, and Lingxi Chen*Nucleic Acids Research, 2026
CNAScope: Pan-Cancer Copy Number Aberration Database with Functional Annotation and Interactive VisualizationXikang Feng†*, Jieyi Zheng†, Sisi Peng†, Anna Jiang†, Ka Ho Ng, Chengshang Lyu, Qiangguo Jin*, and Lingxi Chen*Nucleic Acids Research, 2026Copy number aberrations (CNAs) are critical drivers of genomic diversity in oncology, where recurrent CNAs frequently underlie tumorigenesis. However, existing public resources are limited in their somatic CNA specificity, breadth across multiple data modalities, and support for recurrent CNAs with online functional annotation and interactive visualization. Here, we present CNAScope (https://cna.compbio.com/), a database that curates and functionally annotates over 3,954,361 CNA profiles and 3,946,319 metadata from 810 datasets, 174,464 samples, 3,018,672 single cells, and 764,232 spatial cells/spots, spanning 77 cancer subtypes from eight data sources and 55 cancer initiatives and institutions. CNAScope offers downloadable CNA annotations and interactive visualizations at bin, gene, and pathway term levels, including phylogenetic inference, clustering, dimension reduction, and focal/consensus CNA detection. Users can explore data through interactive heatmaps, phylogenetic trees, embedding plots, CN charts, and focal/consensus plots, or upload and annotate their own CNAs in real time. In all, with its large curated data volume and rich annotation capabilities, CNAScope serves as a vital resource for accelerating cancer research.
@article{CNAScope, title = {CNAScope: Pan-Cancer Copy Number Aberration Database with Functional Annotation and Interactive Visualization}, author = {Feng, Xikang and Zheng, Jieyi and Peng, Sisi and Jiang, Anna and Ng, Ka Ho and Lyu, Chengshang and Jin, Qiangguo and Chen, Lingxi}, journal = {Nucleic Acids Research}, pages = {D1364–D1375}, year = {2026}, publisher = {Oxford University Press}, doi = {10.1093/nar/gkaf1242}, peerreviewed = {true}, member = {Jiang, Anna and Ng, Ka Ho and Lyu, Chengshang}, topic = {CNA, Database, Cancer} } -
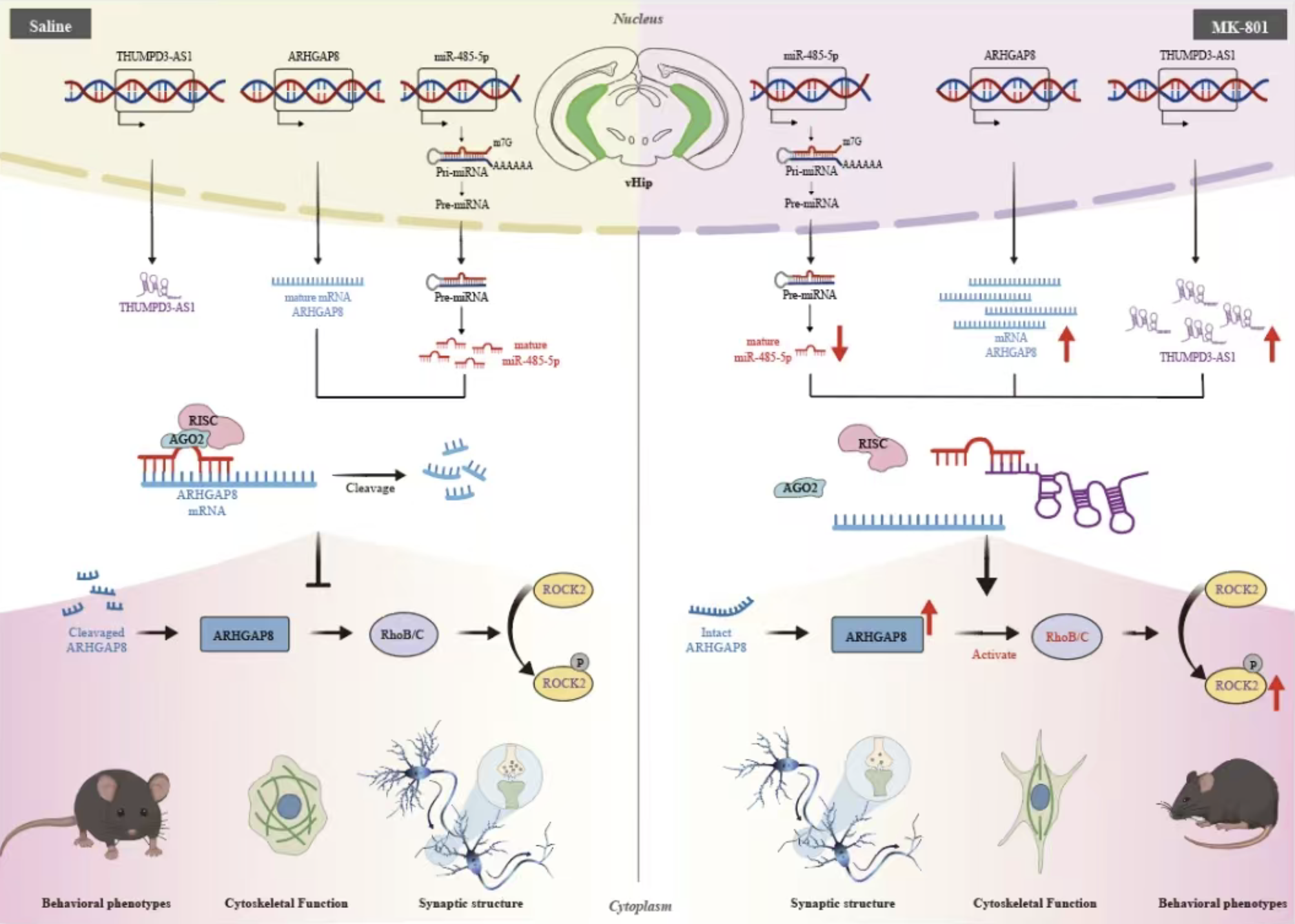 LncRNA THUMPD3-AS1 Regulates Behavioral and Synaptic Structural Abnormalities in Schizophrenia via miR-485-5p and ARHGAP8Xiaojuan Gong, Lingxi Chen, Xin Guo, Anna Jiang, Yayi He, Chunxia Yan, Liang Ma, Jiayang Gao, Jinyu Zhang, and Bao ZhangAdvanced Science, 2025
LncRNA THUMPD3-AS1 Regulates Behavioral and Synaptic Structural Abnormalities in Schizophrenia via miR-485-5p and ARHGAP8Xiaojuan Gong, Lingxi Chen, Xin Guo, Anna Jiang, Yayi He, Chunxia Yan, Liang Ma, Jiayang Gao, Jinyu Zhang, and Bao ZhangAdvanced Science, 2025Abstract Schizophrenia (SCZ) is characterized by synaptic structural deficits, yet how dysregulated noncoding RNAs (ncRNAs) drive these abnormalities remains unknown. Through integrative multilayered analysis of SCZ data from whole transcriptome sequencing (blood samples), GWAS risk loci, and expression data using pipeline ceRNAxis, the THUMPD3-AS1/miR-485-5p/ARHGAP8 axis is identified as a key regulator of synaptic function. Functional validation reveals that THUMPD3-AS1 acts as a competitive endogenous RNA, sequestering miR-485-5p and thereby derepressing ARHGAP8. Despite suppressing RhoA activity, ARHGAP8 enhances ROCK2 activation through RhoB/C-mediated compensatory mechanisms. Hyperactivation of ROCK2 through this noncanonical pathway disrupted actin cytoskeletal remodeling patterns, leading to increased immature dendritic spines and synaptic ultrastructural defects, which are pathological features associated with SCZ. In vivo, ventral hippocampal (vHip) overexpression of miR-485-5p or targeted knockdown of THUMPD3-AS1 rescued MK-801-induced SCZ-like phenotypes (anxiety, cognitive deficits, and social memory impairments) and restored synaptic ultrastructure. Crucially, this regulatory axis is cross-species conservation, with bidirectional expression changes validated in patient-derived blood and vHip tissues of mice. The findings reveal a novel ncRNA-driven pathogenic cascade in SCZ, where dysregulated RhoB/C-ROCK2 signaling, distinct from classical RhoA pathways, mediates synaptic destabilization. This presents a therapeutic axis for precision interventions targeting noncanonical actin cytoskeletal remodeling.
@article{ceRNAxis, author = {Gong, Xiaojuan and Chen, Lingxi and Guo, Xin and Jiang, Anna and He, Yayi and Yan, Chunxia and Ma, Liang and Gao, Jiayang and Zhang, Jinyu and Zhang, Bao}, title = {LncRNA THUMPD3-AS1 Regulates Behavioral and Synaptic Structural Abnormalities in Schizophrenia via miR-485-5p and ARHGAP8}, journal = {Advanced Science}, pages = {e08867}, year = {2025}, doi = {10.1002/advs.202508867}, peerreviewed = {true}, member = {Jiang, Anna}, topic = {ceRNA, Brain Disease} }
- BPformer: An Interpretable Deep Learning Framework for Livestock Breed Proportion AnalysisJinpeng Wang, Shuo Sun, Yaran Zhang, Zhihua Ju, Qiang Jiang, Xiuge Wang, Yao Xiao, Lingxi Chen*, and Jin Ming Huang*Preprint-In Submission, 2025
Introduction: Breed proportion analysis plays a crucial role in cattle genetic resource conservation and breeding improvement. With the rapid development of genomic technologies, breed proportion prediction based on single nucleotide polymorphisms (SNPs) has become a current research hotspot. However, existing methods still face challenges such as insufficient interpretability and the urgent need for feature engineering. Methods: This study developed the BPformer model, which combines convolutional neural networks and self-attention mechanisms, specifically designed for livestock breed proportion prediction. We utilized SNP data from 15 Chinese indigenous cattle breeds and 12 foreign commercial breeds, employing 39,868 high-quality SNPs loci as the gold standard dataset. Dimensionality-reduced datasets were constructed through four feature selection methods (FST, In, BP_AVE, and BP_GRA). The study compared the performance of BPformer against traditional machine learning models (SVR, KNR, and RF) and other deep learning models (MLP, and CNN) on the dimensionality-reduced datasets, while performance evaluation of the three deep learning models was conducted on the gold standard dataset. Results: BPformer outperformed other models across all four detection methods with BBP SNPs = 4,000 and in the gold standard testing scenarios. Through attention mechanism visualization and SHAP value analysis, we identified key SNPs loci that contributed most significantly to the prediction of each breed proportion component, thereby enhancing the model’s interpretability. Conclusion: BPformer effectively addresses the interpretability challenges faced by traditional methods from a modeling perspective and can efficiently capture long-range dependencies among SNPs loci. This provides a powerful tool for Chinese cattle breed resource conservation and genomic selection breeding, which is of great significance for maintaining genetic diversity in Chinese livestock industry.
@article{BPformer, title = {BPformer: An Interpretable Deep Learning Framework for Livestock Breed Proportion Analysis}, author = {Wang, Jinpeng and Sun, Shuo and Zhang, Yaran and Ju, Zhihua and Jiang, Qiang and Wang, Xiuge and Xiao, Yao and Chen, Lingxi and Huang, Jin Ming}, journal = {Preprint-In Submission}, year = {2025}, doi = {10.21203/rs.3.rs-8340493/v1}, peerreviewed = {false}, topic = {Breeding, AI} } -
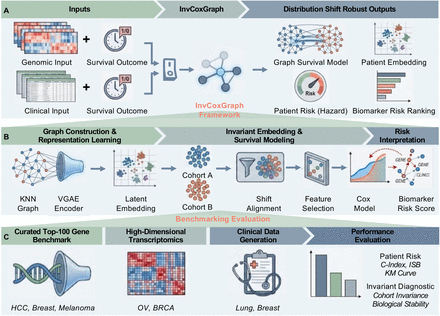 Learning Invariant Graph Representations for Cox Survival Modeling under Distribution ShiftsKa Ho Ng†, Chengshang Lyu†, Anna Jiang, Yinhu Li, and Lingxi Chen*Preprint-In Submission, 2025
Learning Invariant Graph Representations for Cox Survival Modeling under Distribution ShiftsKa Ho Ng†, Chengshang Lyu†, Anna Jiang, Yinhu Li, and Lingxi Chen*Preprint-In Submission, 2025Survival prediction from high-dimensional biomedical data is frequently compromised by distribution shifts across multi-center cohorts, where models trained on specific populations often rely on spurious correlations that fail to generalize to new environments. While recent independence-driven reweighting techniques attempt to mitigate this, they typically treat patients as isolated instances, neglecting the intrinsic topological structures and biological pathways shared within patient populations. To address this limitation, we propose InvGraphCox (Invariant Graph Cox), a novel framework that integrates graph-structured representation learning with robust survival modeling. InvGraphCox constructs a k-nearest-neighbor patient graph to capture local manifold structures and employs a Variational Graph Autoencoder (VGAE) combined with a cohort-wise alignment mechanism to learn low-dimensional patient embeddings that are invariant to site-specific biases. We comprehensively evaluate the framework across three distinct experimental settings: the Curated Top-100 Gene Benchmark for stable biomarker identification, large-scale, high-dimensional transcriptomic datasets (Ovarian and Breast Cancer) for unsupervised representation learning, and clinical datasets (Breast and Lung Cancer) involving mixed-type covariates. Experimental results demonstrate that InvGraphCox consistently outperforms state-of-the-art baselines in terms of discrimination, calibration, and risk stratification, confirming its ability to extract robust, biologically meaningful representations in heterogeneous healthcare settings.
@article{InvCoxGraph, title = {Learning Invariant Graph Representations for Cox Survival Modeling under Distribution Shifts}, author = {Ng, Ka Ho and Lyu, Chengshang and Jiang, Anna and Li, Yinhu and Chen, Lingxi}, journal = {Preprint-In Submission}, year = {2025}, doi = {10.64898/2025.11.30.691365}, peerreviewed = {false}, member = {Jiang, Anna and Ng, Ka Ho and Lyu, Chengshang}, topic = {Survival, AI, Cancer} } -
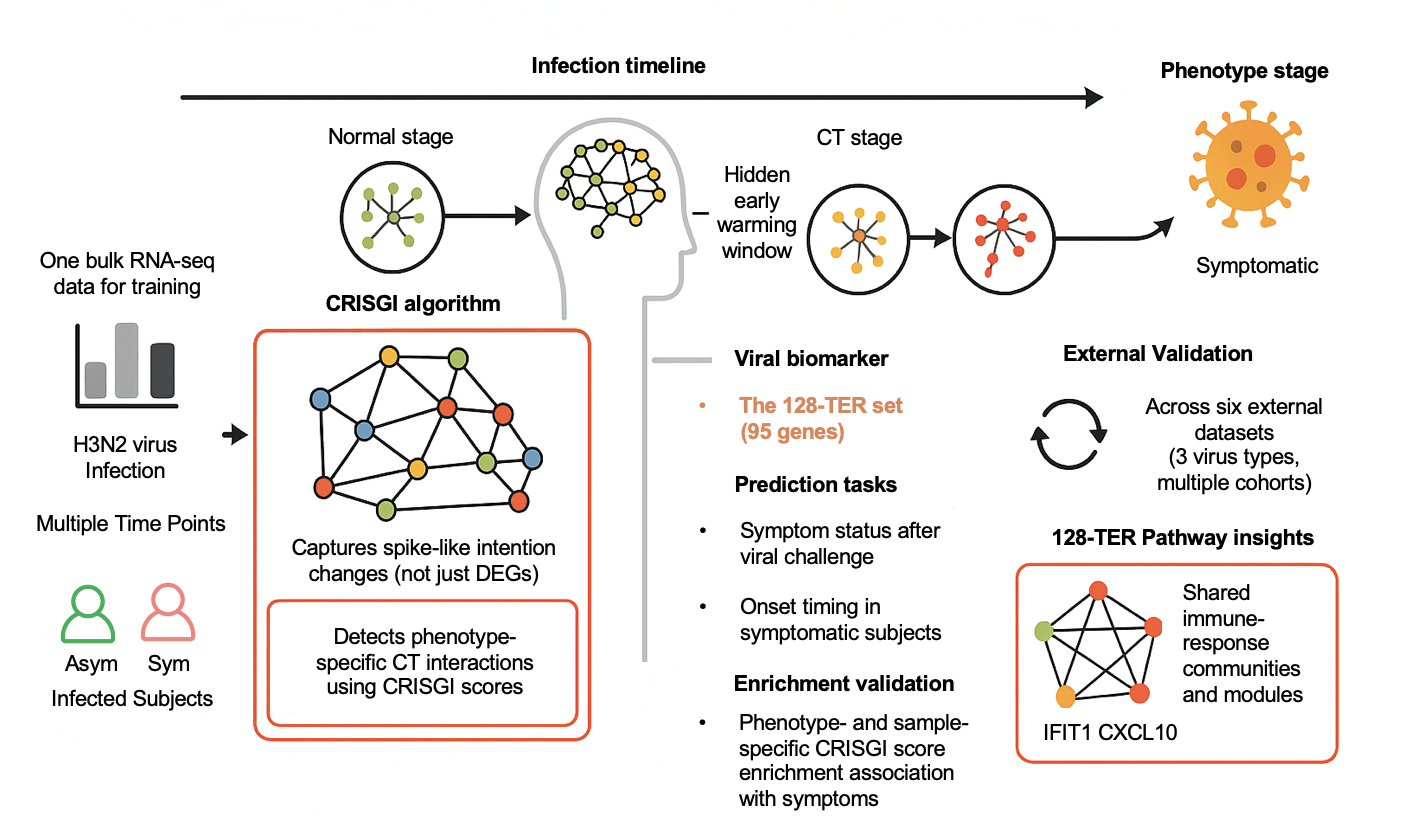 Predicting Early Transitions in Respiratory Virus Infections via Critical Transient Gene InteractionsChengshang Lyu, Anna Jiang, Ka Ho Ng, Xiaoyu Liu, and Lingxi Chen*Preprint-Under Review, 2025
Predicting Early Transitions in Respiratory Virus Infections via Critical Transient Gene InteractionsChengshang Lyu, Anna Jiang, Ka Ho Ng, Xiaoyu Liu, and Lingxi Chen*Preprint-Under Review, 2025Early detection of respiratory virus infections, such as influenza A (H3N2), is critical for timely intervention and disease management. Conventional biomarkers often overlook the complex and dynamic nature of gene regulatory changes, while existing predictive models frequently lack automation and robust external validation. Thus, we present CRISGI (Critical tran-Sient Gene Interaction), a computational framework that detects early-warning signals of infection by identifying dynamic changes in gene-gene interactions—termed critical transient interactions—from bulk RNA-seq data. CRISGI leverages critical transition (CT) theory to capture a GRN’s unstable intermediate state, known as the CT stage, before irreversible phenotypic shifts. Applied to a human challenge study with H3N2, CRISGI identified 128 critical transition edges (128-TER). These were used to train predictive models capable of forecasting symptom status and onset timing. 128-TER was then validated across six temporal transcriptomic datasets involving three respiratory viruses (H3N2, H1N1, HRV). The 128-TER consistently distinguished symptomatic individuals, predicted infection onset, and revealed phenotype-specific enrichment patterns. Notably, CRISGI captured immune-related transitions involving interferon-stimulated genes (e.g., IFIT1, CXCL10), underscoring their role in early host defense. CRISGI advances early-warning biomarker discovery by integrating interaction-level dynamics and predictive modeling. Its reproducibility across viruses highlights shared immune activation pathways, supporting its utility in both research and clinical contexts.
@article{CRISGI, title = {Predicting Early Transitions in Respiratory Virus Infections via Critical Transient Gene Interactions}, author = {Lyu, Chengshang and Jiang, Anna and Ng, Ka Ho and Liu, Xiaoyu and Chen, Lingxi}, journal = {Preprint-Under Review}, year = {2025}, doi = {10.1101/2025.04.18.649619}, peerreviewed = {false}, member = {Jiang, Anna and Ng, Ka Ho and Lyu, Chengshang and Liu, Xiaoyu}, topic = {Critical Transition, GRN, AI, scRNA, ST, Cancer} } -
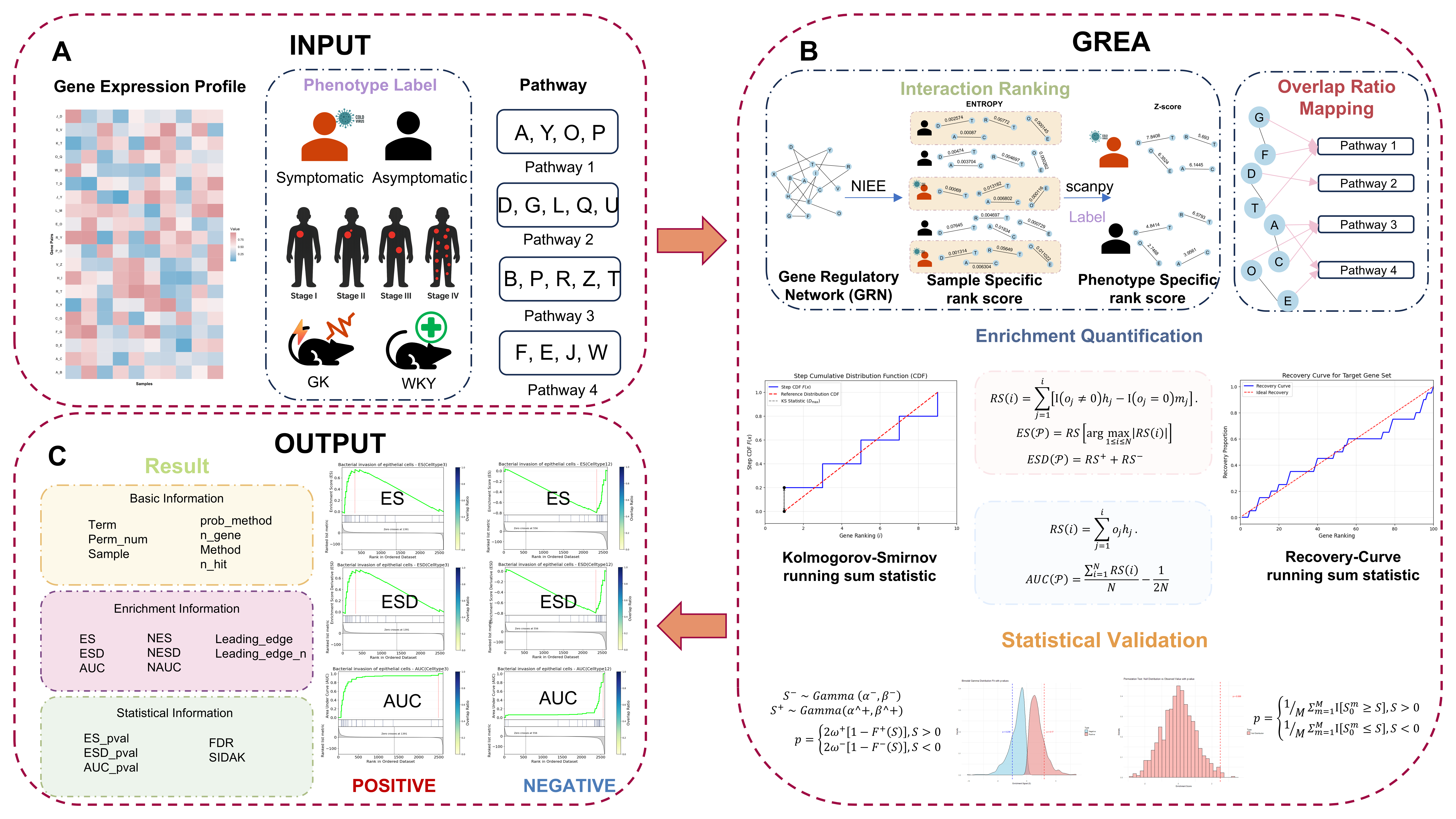 Knowledge-driven annotation for gene interaction enrichment analysisXiaoyu Liu†, Anna Jiang†, Chengshang Lyu, and Lingxi Chen*Preprint-Under Review, 2025
Knowledge-driven annotation for gene interaction enrichment analysisXiaoyu Liu†, Anna Jiang†, Chengshang Lyu, and Lingxi Chen*Preprint-Under Review, 2025Gene Set Enrichment Analysis (GSEA) is a cornerstone for interpreting gene expression data, yet traditional approaches overlook gene interactions by focusing solely on individual genes, limiting their ability to detect subtle or complex pathway signals. To overcome this, we present GREA (Gene Interaction Enrichment Analysis), a novel framework that incorporates gene interaction data into enrichment analysis. GREA replaces the binary gene hit indicator with an interaction overlap ratio, capturing the degree of overlap between gene sets and gene interactions to enhance sensitivity and biological interpretability. It supports three enrichment metrics: Enrichment Score (ES), Enrichment Score Difference (ESD) from a Kolmogorov-Smirnov-based statistic, and Area Under the Curve (AUC) from a recovery curve. GREA evaluates statistical significance using both permutation testing and gamma distribution modeling. Benchmarking on transcriptomic datasets related to respiratory viral infections shows that GREA consistently outperforms existing tools such as blitzGSEA and GSEApy, identifying more relevant pathways with greater stability and reproducibility. By integrating gene interactions into pathway analysis, GREA offers a powerful and flexible tool for uncovering biologically meaningful insights in complex datasets. The source code is available at https://github.com/compbioclub/GREA.
@article{GREA, title = {Knowledge-driven annotation for gene interaction enrichment analysis}, author = {Liu, Xiaoyu and Jiang, Anna and Lyu, Chengshang and Chen, Lingxi}, journal = {Preprint-Under Review}, year = {2025}, doi = {10.1101/2025.04.15.649030}, peerreviewed = {false}, member = {Jiang, Anna and Lyu, Chengshang and Liu, Xiaoyu}, topic = {GRN, scRNA, ST, Cancer} } -
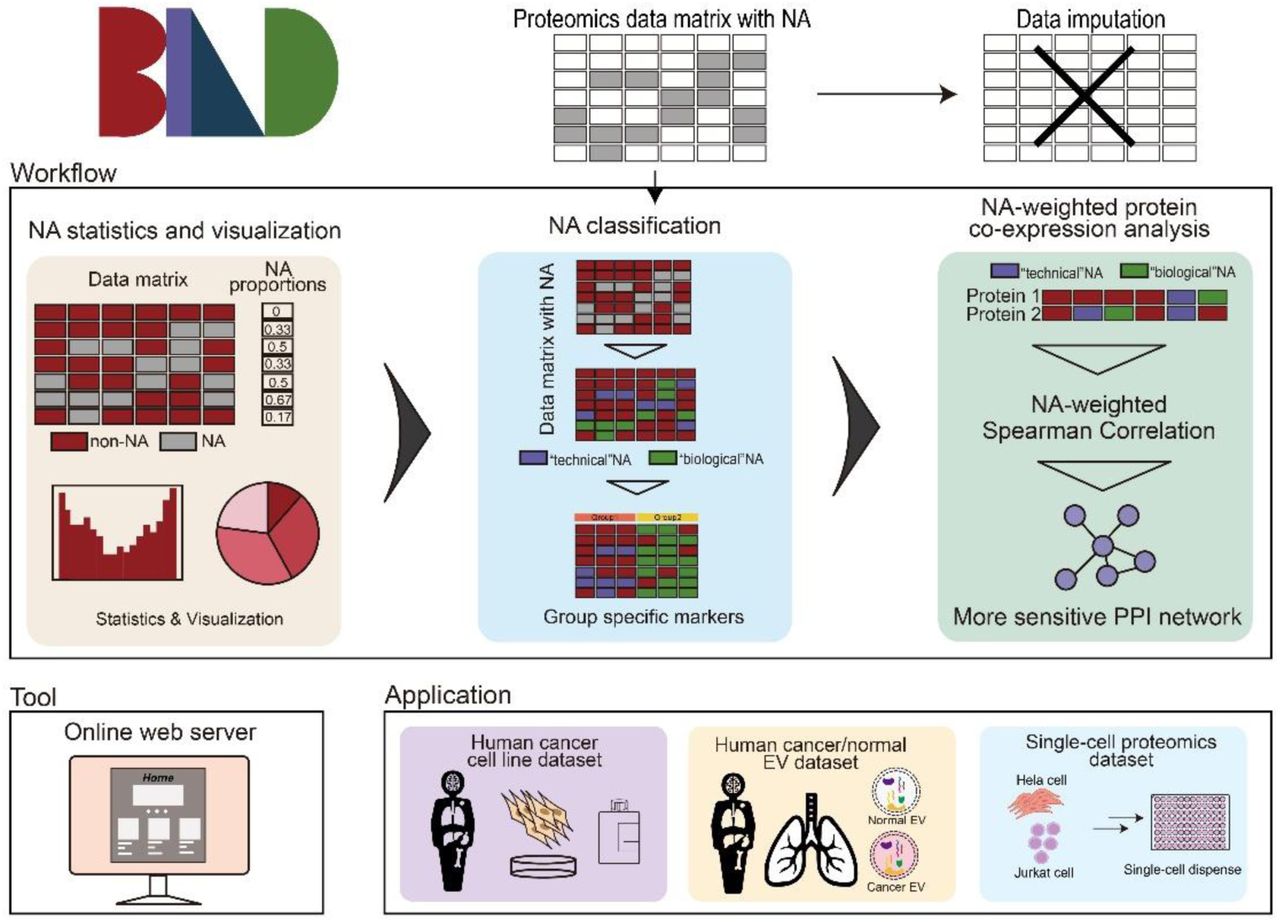 Biologically Informative NA Deconvolution (BIND) excavates hidden features of the proteome from missing values in large-scale datasetsWeiheng Guo†, Wenyi Jin†, Jieyi Zheng†, Yilin Pan, Rui Wang, Jian Zhang*, Xikang Feng*, Lingxi Chen*, and Liang Zhang*Preprint-Under Revision, 2025
Biologically Informative NA Deconvolution (BIND) excavates hidden features of the proteome from missing values in large-scale datasetsWeiheng Guo†, Wenyi Jin†, Jieyi Zheng†, Yilin Pan, Rui Wang, Jian Zhang*, Xikang Feng*, Lingxi Chen*, and Liang Zhang*Preprint-Under Revision, 2025The fast-advancing mass spectrometry and related technologies have greatly extended the depth of coverage in large-scale proteomics studies, including single-cell applications. As sample numbers grow rapidly, it is often challenging to interpret the proteins with missing values that are often presented as “NA” (not available). It could be the evidence of no expression, low expression below the detection threshold, or false negative detection due to technical issues. Existing methods for missing values imputation, while generally useful, rarely consider the non-random NA values that inform biological significance. In the current study, we developed Biologically Informative NA Deconvolution (BIND) that applies an adaptive neighborhood-based modeling to deconvolve the nature of NAs as “biological” (low/no expression) or technical (experimental errors). Applying to multiple cell line datasets and human tissue extracellular vesicle datasets, BIND excavated the NAs that indicated “hallmark absence” of unique proteins. This led to improvements in protein-protein interaction analysis and the identification of novel disease biomarkers. To facilitate its public accessibility, we compiled BIND into a web server that features functional online operations and interactive visualizations. Furthermore, we demonstrated that the BIND server could deconvolve the NAs and improve the analyses of single-cell proteomics datasets. Overall, BIND delineates the biological significance of missing values rather than treating them as a burden, providing a critical perspective for understanding the complex proteome in various biological contexts.
@article{BIND, title = {Biologically Informative NA Deconvolution (BIND) excavates hidden features of the proteome from missing values in large-scale datasets}, author = {Guo, Weiheng and Jin, Wenyi and Zheng, Jieyi and Pan, Yilin and Wang, Rui and Zhang, Jian and Feng, Xikang and Chen, Lingxi and Zhang, Liang}, journal = {Preprint-Under Revision}, year = {2025}, doi = {10.1101/2025.06.19.660508}, peerreviewed = {false}, topic = {Imputation, Proteomics, Cancer} }
{kind=link}
{kind=link}